
Ultimo aggiornamento 19.04.2021
REALIZZATO CON IL CONTRIBUTO NON CONDIZIONANTE DI


L’angioedema da deficit di C1 inibitore (C1-INH) è una malattia rara dovuta alla carenza della proteina plasmatica C1 inibitore. Il deficit di C1-INH può essere dovuto ad un difetto genetico (angioedema ereditario) oppure ad un aumentato consumo della proteina (angioedema acquisito).
L’angioedema ereditario colpisce 1 persona su 50.000. In uno studio nazionale di ITACA su pazienti affetti da angioedema ereditario afferenti ai centri regionali di riferimento è stata calcolata una prevalenza in Italia di 1:64.000.
L’angioedema acquisito è una malattia ancora più rara, non esistono studi sulla prevalenza di tale malattia. In Italia si calcola una prevalenza di circa 1 caso di angioedema acquisito ogni 8 casi di angioedema ereditario.
ANGIOEDEMA EREDITARIO
Nell’angioedema ereditario il deficit di C1 inibitore è dovuto ad una mutazione nel gene del C1-INH. La malattia si trasmette come carattere autosomico dominante. Nella maggior parte dei casi è presente una storia familiare, tuttavia nel 25-30% dei casi la storia familiare è assente. Si parla in questo caso di mutazioni de novo. Sono state riportate in letteratura più di 400 mutazioni diverse. Le mutazioni genetiche determinano più comunemente una ridotta sintesi della proteina C1 inibitore e quindi ridotti livelli plasmatici antigenici e funzionali di C1-INH (angioedema ereditario tipo 1, circa l’85% dei casi). In alcuni tipi particolari di mutazione invece la proteina C1 inibitore viene sintetizzata ma non è funzionante: i valori antigenici di C1-INH nel plasma sono quindi normali mentre i valori funzionali di C1 inibitore sono ridotti (angioedema ereditario tipo 2, circa 15% dei casi). Queste due forme di angioedema ereditario, tipo 1 e tipo 2, sono indistinguibili nella loro presentazione clinica.
MECCANISMI FISIOPATOLOGICI
Il mediatore dei sintomi è la bradichinina che viene liberata in seguito ad attivazione della fase di contatto non controllata dal C1 inibitore. Il C1-INH, infatti, oltre a regolare l’attivazione della via classica e delle lectine del sistema del complemento, è il principale inibitore del sistema di contatto inibendo il Fattore XII attivato e la callicreina plasmatica e svolge anche un ruolo di inibizione della fibrinolisi controllando la formazione di plasmina. In seguito all’attivazione del sistema di contatto (per esempio in seguito a traumi) e alla formazione di FXII attivato, la pre-callicreina viene convertita in callicreina, che clivando il chininogeno ad alto peso molecolare (HMWK) libera la bradichinina (BK) . La plasmina, che si genera dal plasminogeno, agisce potenziando l’attività della callicreina nel liberare BK dal chininogeno ad alto peso molecolare. La bradichinina a sua volta si lega al suo recettore specifico a livello endoteliale determinando un incremento della vaso-permeabilità e quindi la formazione di angioedema (Fig. 1). Nell’angioedema ereditario a causa del deficit di C1-INH i sistemi da esso regolati sono cronicamente attivi o comunque risentono di una certa instabilità. Come conseguenza si ha una maggiore produzione di bradichinina in condizioni basali che aumenta ulteriormente durante un attacco di angioedema.
Figura 1. Ruolo del C1 inibitore nella regolazione del sistema di contatto, fibrinolitico e del complemento. C1-INH = C1 inibitore; fXIIa = Fattore XII attivato; HMWK = high-molecular-weight kininogen; MBL = mannan-binding lectin; MASP2 = mannan-binding-lectin-associated protease 2

FATTORI CHE POSSONO SCATENARE GLI ATTACCHI DI ANGIOEDEMA
Nonostante il deficit di C1 inibitore sia costante gli attacchi di angioedema si verificano in maniera episodica. Esistono dei fattori che possono scatenare le crisi di angioedema come traumi fisici, stress psicologici, interventi chirurgici soprattutto a livello del cavo orale, infezioni, cambiamenti ormonali, assunzione di ACE-inibitori. Spesso, tuttavia, un attacco si presenta senza evidenza di alcun fattore scatenante.
Tabella 1. Fattori che possono scatenare gli attacchi di angioedema. Quando possibile evitare questi fattori scatenanti, per esempio i farmaci (ACE-inibitori ed estrogeni). In caso di interventi chirurgici eseguire prima profilassi con farmaci (vedi profilassi a breve termine)

MANIFESTAZIONI CLINICHE
Da un punto di vista clinico l’angioedema ereditario è caratterizzato da episodi ricorrenti di edema localizzati alla cute, all’addome o alle prime vie aeree. Gli edemi, nella maggior parte dei casi, interessano una singola sede, anche se non sono rari casi di interessamento di più distretti corporei nel corso di un attacco. Gli attacchi si possono manifestare con diversi gradi di intensità, da lievi a moderati a intensi a seconda dell’inabilità a svolgere le normali funzioni della vita quotidiana e lavorativa causata dall’attacco. Gli edemi cutanei possono interessare qualunque parte del corpo e tipicamente si presentano senza prurito; in alcuni casi possono essere associati a dolore soprattutto all’esordio dei sintomi.
Gli edemi alle mani e ai piedi possono causare una temporanea inabilità.
Gli edemi localizzati al volto possono essere disfiguranti (Fig.2).

L’edema della mucosa del tratto gastrointestinale può causare sensazione di gonfiore e dolore addominale intenso, talora associato a vomito e/o diarrea, e può simulare un quadro di addome acuto (Fig. 3). Alcuni pazienti sono stati sottoposti ad interventi di chirurgia addominale non necessari prima di essere correttamente diagnosticati.


Fig. 3. Occlusione intestinale documentata mediante videocapsula
L’edema della glottide (Fig. 4) si manifesta inizialmente come sensazione di corpo estraneo in gola; successivamente possono comparire difficoltà alla deglutizione, cambiamento della voce e infine difficoltà a respirare fino all’arresto respiratorio. L’edema della glottide, pertanto, è da considerarsi un’emergenza medica in quanto può determinare l’ostruzione completa delle alte vie aeree e causare morte per asfissia (se non trattato prontamente con farmaci specifici).
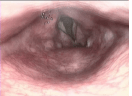
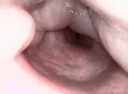
Fig. 4. Edema della glottide
ETÀ DI INSORGENZA DEGLI ATTACCHI
L’età di insorgenza del primo attacco è variabile, con rare segnalazioni di episodi di angioedema già nel periodo perinatale. Nella maggior parte dei pazienti la malattia si presenta per la prima volta durante l’infanzia o nel corso dell’adolescenza. La frequenza degli attacchi aumenta di solito dopo la pubertà.
RITARDO NELLA DIAGNOSI
A causa della rarità di questa malattia e delle caratteriste cliniche simili ad altre forme di angioedema più comuni come gli angioedemi istaminergici i pazienti spesso vengono diagnosticati dopo diversi anni dall’esordio dei sintomi. In Italia si stima un ritardo nella diagnosi maggiore di 10 anni.
VARIABILITÀ NELLA PRESENTAZIONE CLINICA
Il fenotipo di malattia è estremamente variabile da pazienti pauci-sintomatici che presentano rari episodi di angioedema a pazienti che presentano invece frequenti episodi di angioedema (4 o più episodi al mese) e con sintomi severi. Non è chiara la causa di questa variabilità tra i pazienti. Anche nell’ambito della stessa famiglia (quindi in pazienti con la medesima mutazione) si possono rilevare fenotipi di malattia completamente differenti. I sintomi, inoltre, possono variare come frequenza ed intensità anche nello stesso paziente nel corso della vita.
Non è infrequente che gli attacchi di angioedema siano preceduti da prodromi quali stanchezza ed eritema marginato (Fig. 5). Dopo circa 24 ore dalla comparsa dei prodromi si hanno le manifestazioni dell’edema vero e proprio che evolve nelle 24 ore successive, per regredire poi in 2-5 giorni. Nel caso degli edemi laringei con interessamento della glottide l’evoluzione dei sintomi può essere più rapida ed è quindi necessario intervenire prontamente.

IMPATTO SULLA QUALITÀÀ DELLA VITA DEI PAZIENTI
Gli angioedemi regrediscono completamente senza lasciare alcuna sequela. Al di fuori delle crisi di angioedema i pazienti non presentano nessuna limitazione nello svolgimento delle normali attività della vita quotidiana. Tuttavia, in corso di un attacco di angioedema, i pazienti possono essere in una situazione di parziale o completa inabilità nell’esecuzione delle attività della vita sociale e lavorativa. In caso di attacchi frequenti o di intensità grave la malattia può essere invalidante per i pazienti. Inoltre, gli attacchi di angioedema sono spesso imprevedibili. La paura di che si presenti improvvisamente un attacco può quindi causare ulteriori limitazioni nella vita dei pazienti.
ESAMI DI LABORATORIO
Gli esami di laboratorio necessari per porre diagnosi di angioedema ereditario sono il dosaggio antigenico e funzionale del C1 inibitore. Entrambi sono inferiori al 50% in caso di angioedema ereditario tipo 1. Nel caso di angioedema ereditario tipo 2, invece, i valori di C1-INH antigenico sono nella norma mentre i valori di C1-INH funzionale sono inferiori al 50%.
Dal momento che non tutti i laboratori possono eseguire il dosaggio del C1 inibitore, nel sospetto di angioedema ereditario un ottimo test di screening è la misurazione dei livelli plasmatici di C4 che si trovano ridotti nel 96% dei pazienti. Il dosaggio del C4, quindi, pur non essendo un test specifico per l’angioedema ereditario è un test molto sensibile per cui può essere usato come test di screening. Nel caso in cui dovesse risultare normale, infatti, la probabilità di una diagnosi di angioedema ereditario sarebbe molto bassa mentre, nel caso in cui risultasse ridotto, si dovrebbe procedere a dosare il C1 inibitore in quanto la probabilità di un angioedema ereditario sarebbe molto alta. I livelli di C3 e C1q invece sono nella norma. Gli anticorpi anti-C1 inibitore sono di solito assenti e non vengono misurati di routine (nelle forme acquisite di angioedema da carenza di C1-INH, al contrario, il C1q è usualmente ridotto e possono essere presenti questi anticorpi) (Tab 2).
Tab.2. Assetto del complemento nell’angioedema ereditario ed acquisito da carenza di C1-INH

TERAPIA
Lo scopo del trattamento dell’angioedema ereditario è evitare la morte per edema della glottide e ridurre al minimo lo stato di invalidità correlato all’angioedema.
Esistono delle terapie per trattare gli attacchi acuti di angioedema che sono salvavita nel caso di un attacco di edema laringeo e che hanno lo scopo di ridurre la durata e la gravità dei sintomi negli altri tipi di attacco. Esistono poi delle terapie di profilassi per prevenire gli attacchi che hanno lo scopo di ridurre la frequenza e la gravità dei sintomi.
TERAPIA DEGLI ATTACCHI ACUTI/TERAPIA AL BISOGNO
Gli attacchi di angioedema devono essere trattati con i farmaci specifici per questa malattia rara. Gli antistaminici e i cortisonici, usati negli angioedemi istaminergici, non sono efficaci in questa forma di angioedema mediata dalla bradichinina.
I farmaci specifici per l’angioedema ereditario sono (Tab. 3):
- Il concentrato di C1 inibitore (derivato plasmatico o ricombinante) somministrato per via endovenosa.
- L’icatibant, antagonista del recettore della bradichinina, somministrato per via sottocutanea.
I farmaci possono essere utilizzati in qualsiasi tipo di attacco all’inizio dei sintomi. I farmaci, infatti, sono più efficaci nel bloccare la progressione dell’edema e nel ridurre la durata dei sintomi se utilizzati all’inizio dell’attacco. Solitamente un trattamento è sufficiente per risolvere una crisi ma in alcuni casi è necessario un secondo trattamento. Per questo motivo i pazienti devono avere sempre a disposizione almeno due trattamenti per gli attacchi acuti e devono possibilmente essere in grado di auto-somministrarsi i farmaci. In questo modo infatti i pazienti non si devono più recare in ospedale per essere trattati evitando inutili attese, riducendo il tempo dall’inizio dei sintomi alla somministrazione dei farmaci e ottenendo migliori risultati e un miglior controllo della malattia
PROFILASSI A LUNGO TERMINE
Per i pazienti che presentano attacchi di angioedema frequenti o particolarmente gravi e invalidanti deve essere preso in considerazione un trattamento di profilassi a lungo termine che consiste nel trattare i pazienti in maniera continuativa, indipendentemente dalla presenza di un attacco, per prevenire i sintomi (Tab. 3).
I farmaci utilizzati per la terapia di profilassi a lungo termine sono:
- l’acido tranexamico somministrato per via orale
- efficace in una bassa percentuale di pazienti; usato prevalentemente nell’infanzia o in situazioni particolari in cui non sia possibile usare un’altra terapia di profilassi perché controindicata
- la dose può variare da 1,5 gr a 3 gr al giorno
- possono essere presenti effetti indesiderati soprattutto gastrointestinali
- gli androgeno-derivati somministrati per via orale
- sono di solito efficaci nel prevenire gli attacchi di angioedema
- la dose non deve superare i 200 mg die
- mal tollerati soprattutto nelle donne soprattutto in età fertile a causa degli effetti indesiderati come amenorrea, virilizzazione, aumento di peso, dolori muscolari e crampi
- il C1 inibitore derivato plasmatico somministrato per via endovenosa
- iniezioni ripetute di C1 inibitore derivato plasmatico si sono dimostrate efficaci nel ridurre gli attacchi di angioedema
- dose: 1000 U di C1-INH ogni 3-4 giorni
- Lanadelumab (anticorpo monoclonale anti-kallikreina) somministrato s.c.
- ha dimostrato un’ottima efficacia nel ridurre gli attacchi di angioedema
- dose: 1 fiala (300 mg/2 ml) ogni 14 giorni per 6 mesi, quindi rivalutazione clinica per valutare eventuale riduzione ad 1 fiala ogni 28 giorni
- solitamente ben tollerato, sono stati riportati principalmente effetti avversi al sito di iniezione (dolore, bruciore, rush cutaneo) che si risolvono in alcuni minuti
PROFILASSI A BREVE TERMINE
Punta a prevenire gli attacchi in situazioni particolari in cui è molto probabile che il paziente sviluppi un attacco. La profilassi a breve termine è sempre raccomandata in caso di interventi chirurgici sul cavo orale perché possono scatenare un edema laringeo. Il farmaco più utilizzato è il C1 inibitore plasmatico alla dose di 1000 U all’incirca un’ora prima della procedura.
Tab. 3. Farmaci utilizzati per il trattamento dell’angioedema ereditario

ANGIOEDEMA ACQUISITO
Nel caso dell’angioedema acquisito la carenza di C1 inibitore non è dovuta ad un difetto genetico ma è causata dal consumo di tale proteina. L’angioedema acquisito infatti è spesso secondario alla presenza di una malattia linfoproliferativa o autoimmune associata frequentemente alla presenza di autoanticorpi anti-C1 INH.
Da un punto di vista clinico è simile all’angioedema ereditario con episodi recidivanti di edemi che interessano la cute, l’addome o la glottide. Nell’angioedema acquisito sono più frequentI edemi localizzati al volto, alla lingua e all’uvula.
Si differenzia dall’angioedema ereditario per l’assenza di familiarità e per l’insorgenza dei sintomi in età adulta, solitamente dopo i 40 anni di età.
Per quanto riguarda gli esami di laboratorio i valori di C1 inibitore antigenico possono essere normali o ridotti mentre i valori di C1 inibitore funzionale sono sempre ridotti. Come nell’angioedema ereditario i valori di C4 sono ridotti. A differenza dell’angioedema ereditario, nella grande maggioranza dei casi valori di C1q sono ridotti e sono presenti anticorpi anti-C1 inibitore (Tab .2).
Per il trattamento dell’angioedema acquisito si utilizzano gli stessi farmaci utilizzati nell’angioedema ereditario (vedi Fig 6) nonostante non siano mai stati effettuati degli studi clinici con questi farmaci nell’angioedema acquisito. Negli episodi acuti si utilizzano il C1 inibitore derivato plasmatico e l’icatibant. In alcuni pazienti, in presenza di anticorpi anti-C1 inibitore, possono essere necessari dosi di C1 inibitore più elevate oppure infusioni ripetute di C1 inibitore per risolvere una crisi. In questi pazienti l’Icatibant risulta più indicato.
Per quanto riguarda la profilassi vengono utilizzati gli stessi farmaci utilizzati nell’angioedema ereditario. L’acido tranexamico sembra essere più efficace rispetto agli androgeni. Non ci sono ancora dati sull’utilizzo del Lanadelumab in questi pazienti.
Nei pazienti in cui è presente un linfoma (tipicamente in questi pazienti è presente un linfoma marginale splenico), la terapia della malattia può portare ad un miglioramento clinico e alla normalizzazione degli esami di laboratorio.
